
All research conducted at Texas Health Resources (THR) is governed by Federal and State regulations pertaining to human research protections, as well as other Texas Health Resources policies and procedures. The Human Research Protection Program (HRPP) Office is a division under THR’s Research Administration department and additionally reports to Texas Health Resources Corporate Compliance. Texas Health Resources Human Research Protection Program Office (HRPPO) is responsible for ensuring that all human-subject research conducted by THR employees or at a THR hospital or THPG clinic is conducted ethically and in compliance with federal regulations and THR policies
Any Institutional Review Board (IRB) being used for the review and oversight of a study in which THR is engaged must have an appropriate Reliance Agreement in place with THR prior to receiving approval to move forward with the submission of the study with the THR entity. The HRPPO will work directly with each IRB to ensure these are in place.
There are two routes for IRB review at Texas Health Resources, however all studies (both for local IRB review and external/commercial IRB review) must be submitted via eIRB and Velos.
- Local IRB review – The University of Texas Southwestern Medical Center’s (UTSW) IRB shall serve as THR’s local IRB. The application will automatically be routed to THR’s HRPPO for review and approval. You may not begin your study without THR HRPPO approval. You will receive a letter when you are approved. This is in addition to the IRB approval letter.
- External/commercial IRB review – External/commercial IRBs that meet THRs criteria for a reliance IRB may be used. A Reliance Agreement must be in place with the IRB and THR prior to submission of a study to that IRB. The HRPPO will assist getting agreements in place as needed. You may not submit your study to an external IRB without THR HRPPO approval. You will receive a letter when you are approved to submit.
Conflict of Interest Committee
A conflict of interest is a situation in which an researcher’s outside financial interest(s) or obligation(s) (real or perceived) have the potential to bias a research project or cause harm to human subjects participating in a research project.
In collaboration with the Institutional Review Board, the Conflict of Interest (COI) Committee ensures that relationships with outside partners and outside financial interests do not jeopardize the protection of human research subjects or bias the conduct, design or reporting of research. Prior to review of a human research protocol, all study team members listed in the application must submit a Conflict of Interest statement for review. The COI Committee is responsible for reviewing any personal financial interests disclosed by researchers, making determinations about whether those outside financial interests constitute conflicts of interest and make recommendations about how those conflicts of interest can be eliminated, reduced or managed.
Scientific Review Committee
The mission of the Scientific Review Committee (SRC) is to promote research excellence at Texas Health Resources by ensuring that the scientific question being asked is relevant and that the design of a study is appropriate to answer that question. The SRC will review applicable human subject research proposals in which THR is engaged prior to IRB review to ensure that they meet an acceptable standard of scientific rigor and merit.
The SRC will routinely review new intervention protocols to be submitted for IRB review except the following:
- Research approved for federal funding (DoD, DoE, DoJ, EPA, HHS, AHRQ, CDC, FDA, NIH, etc.),
- Research approved for corporation funding utilizing an adequate peer review mechanism, and
- Research that qualifies for exemption from IRB review (QI projects, etc.).
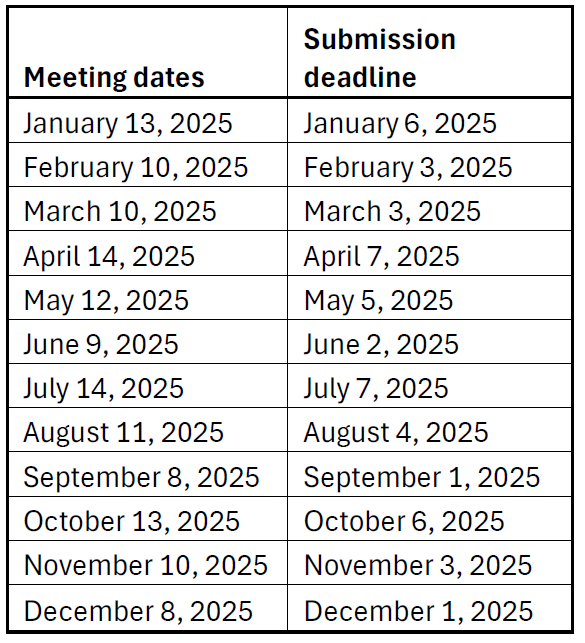
The committee will meet the 2nd Monday of each month and review all projects submitted by the 1st Monday of each month.
How to Determine if Your Study Requires ClinicalTrials.gov Registration
Clinical trials registration and results reporting is required by law for all Applicable Clinical Trials, for clinical trials funded by NIH, and for investigators wishing to publish trial information in an ICMJE journal. The Responsible Party (sponsor) of a clinical trial is the person who initiates the trial. For more information click here.
Helpful Links
https://www.clinicaltrials.gov/policy/fdaaa-801-final-rule
-
THR Human Research Protection Program Office Review Fees
The Human Research Protection Program (HRPP) office provides staff support with conducting research in accordance with federal and institutional regulations and guidelines governing IRB activities. If you have questions or need assistance, please contact us.
Human Research Protection Program (HRPP) fees
Research Administration/HRPP Initial Review Fee (commercial sponsored)
$3,000
Research Administration/HRPP Initial Review Fee (unfunded/non-commercial sponsor)
$1,500
Research Administration/HRPP Initial Review Fee for chart review studies (commercial sponsored)
$1,500
Research Administration/HRPP Initial Review Fee for chart review studies (unfunded/non-commercial sponsor)
$750
Research Administration/HRPP Continuing Review Fee (commercial sponsored)
$1,500
Research Administration/HRPP Continuing Review Fee (unfunded/non-commercial sponsor)
$750
Amendment/Modification (other than administrative amendments)
$600
Please see specific IRB website that your study will be submitted to for that IRB’s fee schedule. Studies submitted to the Texas Health Institutional Review Board prior to December 1, 2019 will be charged the above HRPP fees in lieu of IRB fees.
-
IRB Required Training
Texas Health Resources require that investigators and study staff complete CITI Training modules:
- Human Subject Protections (HSP) training
- Research HIPAA Training
*Renewal of HSP training is done every 3 years from the date of completion/receipt by HRPP Office. Research HIPAA Training is currently completed once.
**As part of the continued collaboration between UTSW and THR, THR investigators and research staff will need to add “University of Texas Southwestern Medical Center” (UTSW) as an Institutional affiliation within their CITI user account profile. This change is effective October 26, 2020 for all new and renewed Human Research Subject Protection training. The UTSW affiliation will allow your completed CITI training to auto populate within electronic submissions for research in the UTSW eIRB system and will assist with streamlining the submission/review process. If you are currently up to date on your training, you will not be required to make this change until time for you to do your training renewal. Per THR policy, CITI training is to be renewed every 3 years.
How to link your CITI account with UTSW:
To make changes to the Institutional affiliation, login into CITI (via http://www.citiprogram.org/) and click “add Institutional Affiliation” on the home page. Then type/choose “University of Texas Southwestern Medical Center” (UTSW) and check the box to agree to Terms of Service and finish the selection. After affiliating with UTSW, you will then be able to view and take the courses under their Institution by clicking the “View Courses” button next to “University of Texas Southwestern Medical Center” on the home page. As you renew you CITI training, you will take the UTSW courses in lieu of the THR CITI courses.
Additionally, as a reminder about adding staff to your studies, Texas Health Resources policies require that if individuals are added to your study after initial approval that are not THR employees or not credentialed at a THR hospital, you must let the THR HRPP office know prior to that individual becoming actively engaged in the research study. Please contact the THR HRPP office and provide the Study’s IRB# (begins with STU-), staff’s name and their institutional affiliation if they have one.
If you have questions or need assistance on either of these items or other study issues, please reach out to the THR HRPP office at HRPP@texashealth.org
Click here to access: CITI Training
CITI Instructions:
Please contact the HRPP office for consideration of training from other sources.
For additional information, please contact HRPP office at HRPP@TexasHealth.org or call 682-236-6746.
-
ETHOS (eIRB) Information
Texas Health Resources uses the University of Texas Southwestern Medical Center (UTSW) eIRB system (ETHOS) for all IRB study submissions, including those to be submitted to an external/commercial IRB.
Accessing the ETHOS (eIRB) system
You will need to obtain a UTSW account to access the ETHOS system.
The Account request form is to be returned to Research Access Requests.
How to Submit a New Protocol Using the Ethos System for THR
Navigating the UTSW ETHOS workspace
Create & Submit a Non-Human and Non-Regulated Research Submission
-
Human Subject Research Regulations and Guidelines
Human Subject Research Regulations and Guidelines
45 CFR 46 is a federal policy, enforced by the U.S. Department of Health and Human Services (HHS), for the protection of human subjects. This policy applies to any human subject research supported by any of the 17 agencies of the federal government that support human subject research. The policy also includes additional protections for vulnerable subjects such as pregnant women, human fetuses and neonates, prisoners and children. This policy is also known as the "Common Rule."
This page provides guidance from the Office for Human Research Protection (OHRP) of the HHS on various topics such as adverse events, certificates of confidentiality, and conflicts of interest.
Categories of Research That May Be Reviewed by the IRB Through an Expedited Review Procedure
This page explains and defines the nine categories of expedited review.
This guidance represents OHRP's current thinking on the reviewing and reporting of unanticipated problems and adverse events. It should be viewed as recommendations unless specific regulatory requirements are cited.
Comparison of FDA and HHS Human Subject Protection Regulations
This guidance discusses the similarities and differences between the U.S. Food and Drug Administration's (FDA) 21 CFR 56 and OHRP's 45 CFR 46.
Studies Using Investigational Drugs, Devices, or Biologics
21 CFR 50, enforced by the FDA, codifies the requirements for informed consent. This regulation is almost identical to the regulations set forth by the DHHS's "Common Rule."
21 CFR 56, enforced by the FDA, codifies the requirements for Institutional Review Boards (IRBs). This regulation is almost identical to the regulations set forth by the HHS's "Common Rule".
21 CFR 312 (IDE studies)
21 CFR 312, enforced by the FDA, describes the procedures and requirements governing the use of investigational new drugs (INDs), including procedures and requirements for the submission to, and review by, the FDA.
21 CFR 812 (IDE studies)
21 CFR 812, enforced by the FDA, encourages the discovery and development of useful devices intended for human use, and establishes an ethical freedom for scientific investigators in their pursuit of this purpose. It provides procedures for the conduct of clinical investigations of devices. An investigational device exemption (IDE) permits a device, that would otherwise be required to comply with performance standards or to have premarket approvals, to be shipped lawfully for the purpose of conducting investigations of that device.
FDA Guidances, Information Sheets, and Notices
These guidances and information sheets represent the FDA's current guidance on good clinical practice (GCP) and the conduct of clinical trials.
FDA Guidance for Clinical Investigators, Sponsors, and IRBs
Adverse Event Reporting to IRBs -- Improving Human Subject Protection (issued January 2009).
FDA Guidance for Sponsors, IRBs, Clinical Investigators, and FDA Staff
Informed Consent for In Vitro Diagnostic Device Studies Using Leftover Human Specimens that Are Not Individually Identifiable (issued April 2006).
HIPAA Regulations
45 CFR 160, 162 and 164, enforced by the Office for Civil Rights, is also known as the HIPAA Privacy Rule. It establishes national standards to protect individuals' medical records and other personal health information and applies to health plans, health care clearinghouses, and those health care providers that conduct certain health care transactions electronically.
Conflict of Interest (COI) Regulations & Guidelines
Food and Drug Administration (FDA) Regulations
This guidance represents the Food and Drug Administration's current thinking on this topic.
Public Health Service (PHS) Regulations
Association of American Medical Colleges (AAMC) Guidance
Policy and Guidelines for the Oversight of Individual Financial Interests in Human Subjects Research.
Other IRB-Related Guidance
The Belmont Report was created as a result of the 40-year U.S. Public Health Service Syphilis Study at Tuskegee, in which syphilis subjects were denied treatment for their disease. This legislation, which was passed in 1974, created regulations to protect human subjects and created a National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research to examine ethical issues related to human subject research.
The Nuremburg Code was created as a result of the atrocities committed against humans during World War II. It addressed the significance of obtaining informed consent, of ensuring that this consent was voluntary and of ensuring that any individual "who initiates, directs, or engages in the experiment" bears responsibility for the quality of consent.
The significances of the Nuremberg Code were further articulated and expanded in the Declaration of Helsinki, which was originally set forth in 1964. The Declaration's significance was that it called for prior approval and ongoing monitoring of research by independent ethical review committees.
-
Not Human Subjects Research
-
Federalwide Assurance - Statement of Compliance
Texas Health Resources holds a Federalwide Assurance FWA00013095.
Information relating to this assurance, including the current expiration date, can be viewed on OHRP's website.
Click here to view Texas Health Resources' Documentation of FWA Membership.
Statement of Compliance
-
Emergency Use of a Test Article
Background Information
The emergency use of an investigational drug, device or biologic under FDA regulations at 21 CFR 56.104(c) permits the emergency use of an investigational drug, device or biologic on a one-time basis per Institution without IRB review and approval prior to its use (but the emergency use must be reported to the IRB within 5 working days from the date of use). Contact the THR HRPP office (214-682-6746 or HRPP@texashealth.org) for assistance with questions on completing Emergency Use Forms.
Note: Each of the Texas Health Resources entities and affiliated facilities are considered to be an Institution. Texas Health Resources uses the University of Texas Southwestern Medical Center IRB (UTSW IRB) to review requests for Emergency Use of a Test Article and for Compassionate Use/Expanded Access requests. To submit a request for review to the UTSW IRB, please complete the Individual Patient Treatment form. For Question 4 on the form, select “Non-Research, Treatment Protocols” (this choice includes Emergency Use and Compassionate Use/Expanded Access Requests). Please ensure Question 7a is marked Yes (to designate request is taking place at a Texas Health Resources facility). Please upload any supplemental documents in Question 6.5. Hit the Submit button to route the request to the UTSW IRB office for review.
The FDA acknowledges that it would be inappropriate to deny emergency treatment to a second individual if the only obstacle is that the IRB has not had sufficient time to convene a meeting to review the issue ["Emergency Use of an Investigational Drug or Biologic," FDA Information Sheet, 1998 Update: Click here ].
Data collected from the patient who received emergency treatment cannot be considered research, as defined by the Department of Health and Human Services (DHHS) regulations. Therefore it cannot be used with data for a research study.
Data collected from the patient is considered research under FDA regulations and can be aggregated with other research data, provided that research is not subject to DHHS regulations.
The FDA may require data from an emergency use of a test article in a life-threatening situation to be reported in a marketing application. Therefore, a HIPAA/Privacy authorization should be signed as well.
-
Texas Health Research Policies
-
Report Non-Compliance
What is non-compliance?
Non-compliance is any action or activity associated with the conduct or oversight of research involving human subjects that fails to comply with the research plan as approved by the IRB or federal regulations or institutional policies governing such research. Non-compliance may range from minor to serious, be unintentional or willful, and may occur once or several times.
How do I report non-compliance?
You may contact the Texas Health Resources System Compliance Hotline at 1-800-381-4728 or SystemCompliance@TexasHealth.org
The hotline is a toll-free line dedicated to the confidential discussion of questions and concerns related to laws, regulations, or company policies. If you choose to remain anonymous your identity will be protected to the extent possible or allowed by the law.
How do I avoid non-compliance?
- Know and Comply with company polices
- Complete the required training and education
- Report suspected compliance concerns
- Remember you will be held accountable for complying with policies
Got Questions? Contact the Texas Health Resources Human Research Protection Program (HRPP) Office.
Texas Health Resources HRPP Office
8440 Walnut Hill Lane, Suite 220
Dallas, Texas 75231
Phone: 682-236-6746
Email: HRPP@TexasHealth.org
-
Forms and Templates
FORMS:
- Conflict of Interest Disclosure (COI) - A separate COI form must be completed for each study.
- Entity Reviewer Approval Form
- UTSW Account Request Form
- Study Questionnaire (Required for all studies involving THR facilities and/or staff)
- Research Protocol Template
Additional UTSW Forms and Templates
-
Helpful Resources
Texas Health Resources Code of Business Ethics
This links you to the Texas Health "Code of Business Ethics" handbook.
Language Access Services & Disability Support Services Toolkit
A toolkit created by Texas Health Diversity and Inclusion to show services offered, such as, language interpreters, sign language interpreters, and other translation services.
Links Outside Texas Health Resources
A U.S. government agency within the U.S. Department of Health and Human Service (HHS), the FDA enforces laws on the manufacturing, testing and use of drugs and medical devices.
National Institutes of Health (NIH)
A U.S. Public Health Service (PHS) entity within the DHHS, the NIH serves as the federal government's primary agency for advancing knowledge in biomedical and behavioral sciences in order to understand and treat human diseases.
Department of Health and Human Services
A U.S. government agency, the HHS was established to protect the health of the U.S. population. Its mission specifically includes the protection of all humans participating in clinical research.
Office for Human Research Protections (OHRP)
A U.S. government agency within the HHS, the OHRP helps ensure the protection of humans participating in clinical research.
The National Library of Medicine is the world's largest medical library. It collects materials and provides information and research services in all areas of biomedicine and health care.
Guidance for IRBs, Clinical Investigators, and Sponsors
This is an FDA Web site dedicated to guiding investigators and sponsors in issues related to subjects such as recruitment, charging for investigational products and informed consent..
This is an FDA Web site dedicated to helping investigators, sponsors, and contract research organizations that conduct clinical studies on investigational new drugs comply with U.S. law and regulations covering good clinical practice.
Case reports are not considered research and as such do not require IRB review/approval. However, THR does require the patient to sign a release form.
If you are a physician, please use the following English/Spanish form for your patient.
If you are a hospital employee, please use the following English/Spanish form for your patient.A case report is a detailed report of the diagnosis, treatment, response to treatment, and follow-up after treatment of a individual patient. A case series is a group of case reports involving patients who were given similar treatment. When information on more than three patients is included, the case series is considered to be a systematic investigation designed to contribute to generalizable knowledge (i.e. research) and submission is required to the IRB.
-
FAQs
What is an IRB?
The Institutional Review Board (IRB) is a committee established under federal regulations that reviews human subject research to assure that the rights and welfare of human subjects are adequately protected. It oversees institutional compliance with all federal, state, and local regulations; in addition to Texas Health Resources policies.
What kinds of research studies require IRB approval?
All research involving human subjects or their identified protected health information must be submitted to the IRB for review. A human subject is an individual from whom an investigator conducting research obtains data through intervention or interaction, or identifiable private information. Click here to learn more.
How do I determine if my project requires IRB approval?
You should submit your project to the IRB if it involves human subjects or their identifiable data. Click here to view a tool designed to assist in determining if IRB approval is needed. If you have questions please contact the IRB office.
What is FDA Research?
FDA research is research funded by the U.S. Food and Drug Administration. Click here to learn more.
What kinds of changes to my research require an amendment?
These types of changes require an amendment to be submitted to the IRB. Do not implement the change until you receive an approval letter from the IRB.
Examples of changes to a study include but are not limited to:
- Changes to the protocol
- Changes to the consent form
- Changes to surveys/ questionnaires
- Changes to recruiting materials
- Addition or removal of a stipend
- Changes in study personnel
- Updated investigator's brochure
- Changes in conflict of interest
- Changes to the sample size
- Addition or deletion of sites
- Changes to inclusion/ exclusion criteria
- Revision to data and safety monitoring plan
- Requesting additional funding
- Other changes as determined by the IRB
Can my study be expedited?
Expedited review is a type of IRB review for studies that a) pose no more than a minimal risk to subjects, and b) meet at least one expedited review category. The study will be reviewed by one voting board member instead of the full board. Click here to view the expedited categories.
Is my study exempt from IRB review?
For your study to be exempt from review it must fit into one of the exempt categories. Click here to view the requirements for exemption.
What is a Limited Data Set (LDS)?
Click here to view information on LDS.
What is Protected Health Information (PHI)?
-
Contact the HRPP Office
HRPP Office
Main 682-236-6746
HRPP@TexasHealth.orgTexas Health Research & Education Institute
8440 Walnut Hill Lane, Ste. 220
Dallas, TX 75231If you have a concern or want to report an issue and wish to remain anonymous, you may contact the Texas Health Resources System Compliance Hotline at 1-800-381-4728 or SystemCompliance@TexasHealth.org